Britain’s leading role in evaluating new medicines for sale to patients across the EU has collapsed with no more work coming from Europe because of Brexit, it has emerged.
The decision by the European Medicines Agency to cut Britain out of its contracts seven months ahead of Brexit is a devastating blow to British pharmaceutical companies already reeling from the loss of the EMA’s HQ in London and with it 900 jobs.
All drugs sold in Europe have to go through a lengthy EMA authorisation process before use by health services, and the Medicines & Healthcare products Regulatory Agency (MHRA) in Britain has built up a leading role in this work, with 20-30% of all assessments in the EU.
In a devastating second blow, existing contracts with the MHRA are also being reallocated to bloc members.
Martin McKee, the professor of European health at the London School of Hygiene and Tropical Medicine, who has given evidence to select committees about Brexit, said it was a disaster for the MHRA, which had about £14m a year from the EMA.
The head of the Association of British Pharmaceutical Industry said it was akin to watching a “British success story” being broken up.
Mike Thompson, the chief executive of the association, said: “Clearly we’ve all been incredibly proud of the MHRA’s role over the last few years. They’d established themselves as one of the most respected regulators across all of Europe and industry. It’s been a British success story.”
The EMA said that because of the long lead-time involved in assessing medicines it could no longer award the lead contracts to British people since there was no guarantee they would be part of the EU after March 2019.
It is understood the MHRA bid for 36 EMA contracts this year but were only awarded two, and these were for drugs for which evaluation had already begun.
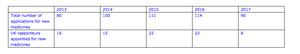
The situation is a stark contrast to 2016 when the UK was the lead assessor, known as the rapporteur, on 22 applications, and was joint lead or co-rapporteur on 19 multinational applications. This made it the number one in Europe, with Germany’s regulator behind with 22 lead contracts but only 12 co-contracts.
The EMA now also requires all existing drugs assessors to transfer their personal knowledge of their specialist fields to counterparts in a European member state.
“You might have been working on a cancer drug for decades and built up so much expertise and you are the absolute specialist in your field and now have to transfer all your knowledge to someone else. It must be like handing over your baby,” said one source in the EMA.
Thompson suggested the removal of the MHRA from the approvals system was the EU’s loss. The MHRA did one-third of all the manufacturing inspections and in terms of patient safety they had picked up one third of all “adverse events”. He said: “This is a pre-eminent regulator. As part of the withdrawal agreement the UK regulator … they will just be an observer in that system.”
McKee said: “The MHRA has benefited enormously from its close links with the EMA. The fracturing of those links will impact severely. on its budget, much now from the EMA, and its ability to attract and retain skilled staff.”
The loss of these valuable specialist contracts underlines the punishing impact Brexit is having on services that supports the pharma industry in Britain.
The EMA has already started its move from Britain to its new headquarters in Amsterdam. It employs 900 staff in its Canary Wharf offices, in London, and 84 have already relocated to the Dutch capital, the EMA said. It expected that about 300 of its staff would be unable to relocate and have to find new jobs because of Brexit.
The ABPI said the change in approvals meant a huge cost to the medicines companies.
Thompson said: “Companies are having to build extra laboratories to try to prepare to batch-release medicines made in the UK on the continent. That’s a huge cost for us. Hundreds of millions of pounds that we’re having to spend that frankly we’d rather spend on researching new medicines. We have no choice because we have to ensure what we do is legal. It’s probably wasted money in the end.
“We regret that, because the MHRA is a highly respected agency. We’d hope that could be resolved as soon as possible. Ultimately it will be difficult for them to hang on to the capability that they have spent many years building up.”
The MHRA said it hoped its relationship with the EMA could be salvaged in negotiations.
An MHRA statement said: “We want to retain a close working partnership with the EU to ensure patients continue to have timely access to safe medicines and medical devices. This involves us making sure our regulators continue to work together, as they do with regulators internationally, and we would like to explore with the EU the terms on which the UK could continue to participate in the EMA.”




