It has now been over six months since the FDA authorized Paxlovid in December 2021 “for the treatment of mild-to-moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing who are also at high risk for progression to severe COVID-19.”
At the time of authorisation, the United States federal government had already purchased 10 million doses of the drug. That number soon doubled to 20 million. It’s unclear how many of those doses have been administered, but according to numbers from Pfizer, usage ramped up steadily between February and April to 80,000 doses per week.
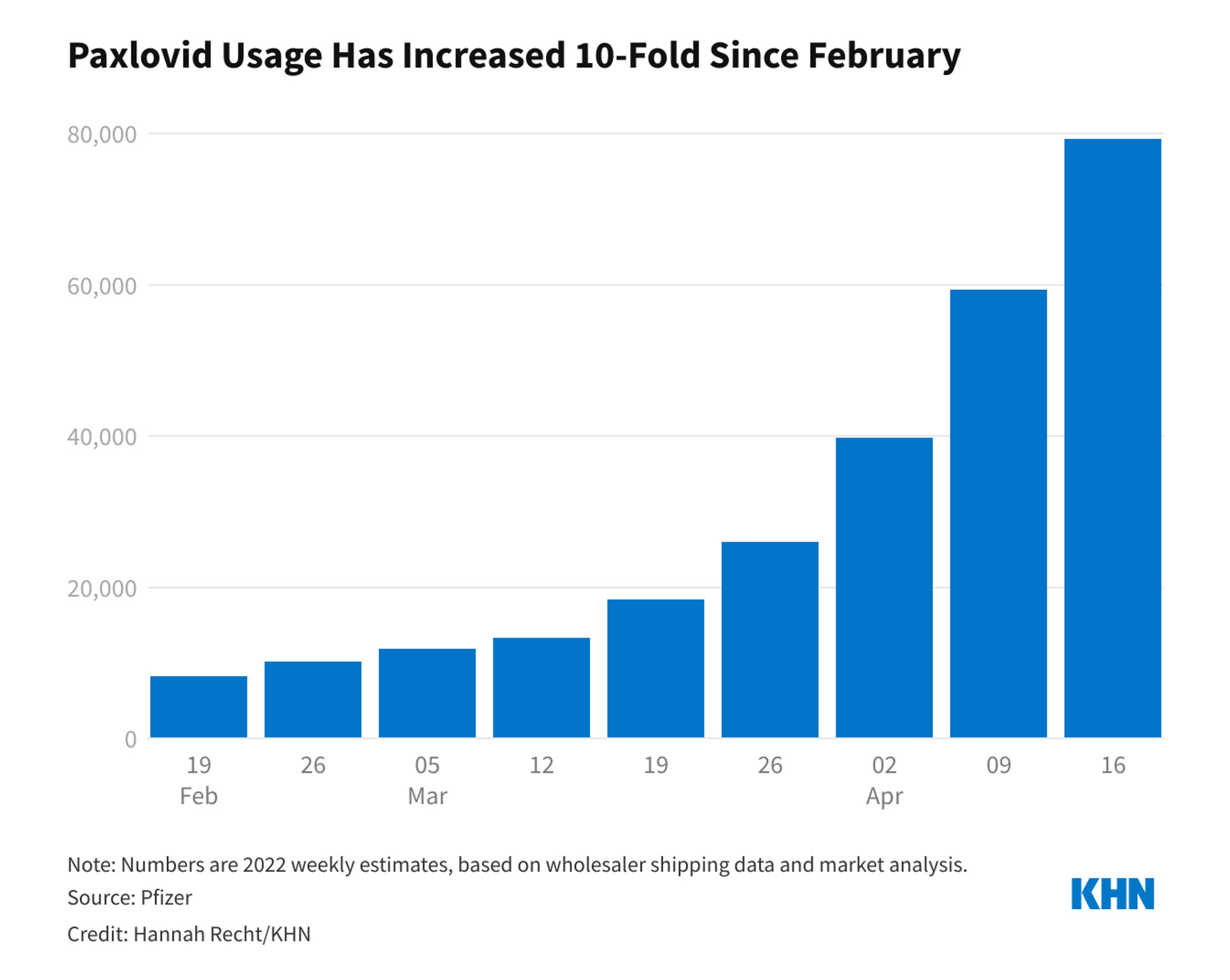
As COVID-19 cases rise once again and the federal government gears up to increase distribution of Paxlovid, we thought we’d take this opportunity to review information about the drug and share new information that has come to light since it was first approved.
What is Paxlovid and how was it developed?
Paxlovid was the first oral antiviral the FDA authorized to treat COVID-19. Not only is it effective but it can be taken at home, which is much more convenient than getting an IV infusion. Paxlovid is used to treat adults and children over 12 years old with mild-to-moderate coronavirus disease who are at high risk for progression to severe disease. Developed by Pfizer, Paxlovid was granted emergency use authorization (EUA) to treat COVID-19 patients based on the EPIC-HR study which showed it reduced the risk of hospitalization or death by almost 90 percent.
Paxlovid can be prescribed for anyone age 12 and older who weighs at least 88 pounds. The patient must have tested positive for COVID-19 and be considered high risk to develop severe COVID-19. High-risk patients have certain medical conditions that may predispose them to progression to severe disease. It is taken for five days, and treatment must be started within the first five days of when symptoms first appeared. Paxlovid consists of two different drugs, nirmatrelvir 150mg tablets co-packaged with ritonavir 100mg tablets. Nirmatrelvir 300mg (two tablets) is taken with ritonavir 100mg twice a day for five days.
How does the drug work?
The active drug is nirmatrelvir, a protease inhibitor, which helps keep the SARS-CoV-2 protein from replicating. SARS-CoV-2-3CL protease is an enzyme COVID-19 needs to replicate and spread. Ritonavir is another protease inhibitor, but its primary function is to keep nirmatrelvir in the body longer so that it can do its thing. Ritonavir has been around for decades and was originally approved to treat HIV.
What are its side effects?
Paxlovid seems well tolerated for now, although studies are ongoing. The drug has been in the news recently because of incidents of dysgeusia — a bad, metallic taste in the mouth. In clinical trials, 5.6 percent of patients who received Paxlovid experienced dysgeusia.
While very unpleasant, this side effect is temporary and not cause for concern. It’s a much greater issue with HIV patients, who take ritonavir for a longer period of time. There isn’t much patients can do about the taste other than wait it out.
Other possible side effects include allergic reactions, liver problems, diarrhea, high blood pressure and muscle aches. Paxlovid interacts with a long list of medications which may cause serious complications, so physicians should have a complete list of medications the patient is on.
How effective is Paxlovid?
Paxlovid is highly effective for acute COVID-19 as it reduces the risk of hospitalization by nearly 90 percent. However, there are still many unanswered questions about how effective the drug is in certain scenarios, as additional studies have not been published since emergency use authorization was granted.
A major blind spot in the data is how effective Paxlovid is at treating vaccinated patients who contract COVID-19. Since the EUA trial only focused on unvaccinated adults, we still are unable to quantify how effective the drug is for individuals who have had at least one shot, are fully vaccinated or have been boosted.
Finally, long COVID has been an area where there hasn’t been much advance in terms of remedies. Paxlovid is no exception as it has not been studied to treat long COVID.
What is “Paxlovid Rebound”?
There have been reports of “Paxlovid rebound” where patients that have taken Paxlovid, may see a recurrence of symptoms after a brief recovery. In addition, researchers have found in some cases, those with rebound infection can be contagious. Limited information from case reports seem to show these rebound infections have resulted in only mild illness thus far. The CDC has issued a Health Alert Network Health Advisory to warn of the potential risk of COVID-19 recurrence after Paxlovid treatment and advises patients to isolate for at least five days after recurrence.
Although preliminary trial data from Pfizer found rebound in 1 to 2 percent of patients, it occurred in both placebo and treatment groups. The extent of how often “Paxlovid rebound” occurs is unknown at this point as we’re not even 100 percent sure this rebound is caused by the drug. Pfizer will continue to monitor data from ongoing clinical trials and post-authorization safety surveillance. The CDC continues to recommend Paxlovid for early-stage treatment of mild to moderate COVID-19 in people that are at high risk for progression to severe disease.
How can people in the US access Paxlovid and what is the cost?
The federal government oversees the allocation to state health departments for distribution to pharmacies, hospitals, urgent cares and other entities. Paxlovid is available by prescription from a healthcare provider and is available at no cost to the patient during the COVID-19 public health emergency.
Are changes needed to make Paxlovid more widely available?
Since February, supply has kept up with demand. Although there may be an assumption that there is not enough of this drug around, I do not believe availability is an issue thus far as recent Health and Human Services data shows more than half of the doses of Paxlovid ordered are sitting on shelves not being used.
There may be different reasons for this underutilization. The omicron variant does not seem to be as severe as previous strains, so some might think they don’t need a drug like Paxlovid since they don’t believe their mild symptoms will get much worse. Lack of advertisement probably contributes to the lack of awareness of treatments such as Paxlovid. Pfizer cannot advertise this treatment since Paxlovid is only authorized for emergency use and not available on the commercial market. It may also be hard to know where Paxlovid is available.
The U.S. Department of Health and Human Services Office of the Assistant Secretary for Preparedness and Response has created a COVID-19 Therapeutics Locator website, but not many people may know about it. I’m not sure if policy changes are needed at this point, but a plan to educate the masses on Paxlovid and how to get the drug may help.
Rob Louie is EVP, Clinical Services at RemedyOne, a formulary and rebate optimization and part of Goodroot, a community of companies dedicated to reinventing healthcare one system at a time. He has more than 30 years of experience as a pharmacist.
https://pharmaphorum.com/market-access-2/six-months-after-authorization-what-do-we-know-about-paxlovid/