My career has really been around clinical research in obesity and diabetes. I was part of the Diabetes Prevention Program, theLook AHEAD study, and thePOUNDS LOST study. I helped develop theDASH diet. All of these studies focused on trying to help patients lose weight so that we can better treat these chronic diseases that are filling our offices.
I have to say, I don't think I was ever really successful in translating what I knew from those clinical research studies into practice. There was always this resistance of primary care practitioners in trying to be effective in weight management. I didn't really have any tools to give them that would work in a modern practice. Primary care practitioners today are very time-pressured. They're seeing patients at these 15-minute intervals, so I need to give them tools that work.
I'm very hopeful about some of the newer medications that are coming out. I think this is likely to change our approach to diabetes management and to management of all chronic diseases. I think we're going to take a much more weight-centric approach.
Some of our newer medications are targeting glucagon-like peptide 1 (GLP-1). GLP-1 and gastric inhibitory polypeptide (GIP) are incretin hormones that improve glycemia and also have an effect on weight. Some of these newer medications are leading to dramatic improvements in weight loss.
I feel like our doctors are finally going to have some tools that are going to help them. Believe me, this has been a long, hard struggle for me. I've been involved in the development of many medications — some of which succeeded and some of which failed — around weight management. I kissed a lot of frogs. Finally, I feel like I'm getting some princes.
Just having effective medications is not enough. Our doctors need to understand how to use these medications and to provide weight management instructions to their patients. Even more important, we have to be able to get these medications into the patients' bodies.
One of the problems is that our payers — our employer-based insurance companies our federal insurance companies — don't always reimburse for these really incredible scientific achievements in drug discovery, and that's especially true around obesity medications.
We have to solve the problem of drug pricing in America if we're going to be able to get these really great drugs into our patients' bodies so that we can make weight management the driver of all these chronic diseases, and so that we can make weight management central to chronic disease management.
Donna H. Ryan, MDis Past President of the World Obesity Federation and professor Emerita at Pennington Biomedical in Baton Rouge, LA, where she directed clinical research for 22 years. Her own research includes participation on the teams that developed and executed the DASH (Dietary Approaches to Stop Hypertension), POUNDS Lost, DPP (Diabetes Prevention Program) and Look AHEAD Studies. Dr. Ryans continuing research interests focus on translation of effective weight management into primary care practices.
Cruise lines face regulation not just from the country where their ships sail from, but also from the countries where they make port stops. That has led to a challenging array of covid protocols based on which ports of call a ship visits.
That makes communication with passengers a challenge. In the early days of Royal Caribbean International (RCL) - Get Royal Caribbean Group Report, Carnival Cruise Lines (CCL) - Get Carnival Corporation Report, Norwegian Cruise Lines (NCLH) - Get Norwegian Cruise Line Holdings Ltd. Report, and other cruise lines returning from their pandemic shutdown rules were constantly shifting. Mask requirements when onboard kept shifting and vaccination rules took a while to settle down.
That was made more complicated by the fact that many of the countries the cruise lines visited had varying rules. Some ports required families with unvaccinated kids to only take cruise line sponsored excursions while others had mask rules that were more strict than the ones on the ship. In addition, some ports required an actual health visa for anyone hoping to get off.
Adding the requirement created an added level of confusion for passengers. There was one set of rules and protocols to get on the ship and another (maybe more than one other) set for certain ports. In some cases, this involved filing paperwork before leaving home and there was often an added cost.
Now, one frequent cruise destination has dropped a key covid rule.
Daniel Kline/TheStreet
Bahamas Ends a Key Covid Rule
In the early days of the pandemic, Royal Caribbean used Nassau, Bahamas as the home port for Adventure of the Seas. That allowed people from the United States to fly to the island nation and board the ship for a sailing similar to the ones that usually left from Florida home ports.
Taking that trip required not just producing a negative covid test but also applying for a Bahamas health visa. The health visa was not a requirement once ships started sailing from the U.S. again for people planning to get off for a port day in the Bahamas. Pre-visit covid testing was still required, but as long as cruise ship passengers were vaccinated, they did not need the health visa.
Paul Verdin, VP of consulting and analytics, and Andreas Hadjivasiliou, managing analyst at Evaluate, tell us about the company’s Orphan Drug Report 2022, which highlights activities within the orphan drug market and forecasts what the future of the industry holds based on data.
Evaluate provides commercial intelligence to the pharma and life sciences sectors through annual publications. They recently released their yearly report on orphan drugs, offering snippets of what’s happening in the rare disease drug market.
“We have a lot of data about pharmaceutical pipelines, sales in the market, and, crucially, about the forecasts and how the market might evolve, which is all derived from sell-side equity analysts,” Verdin states.
“Orphan drugs and rare diseases are a big part of the picture for pharma.”
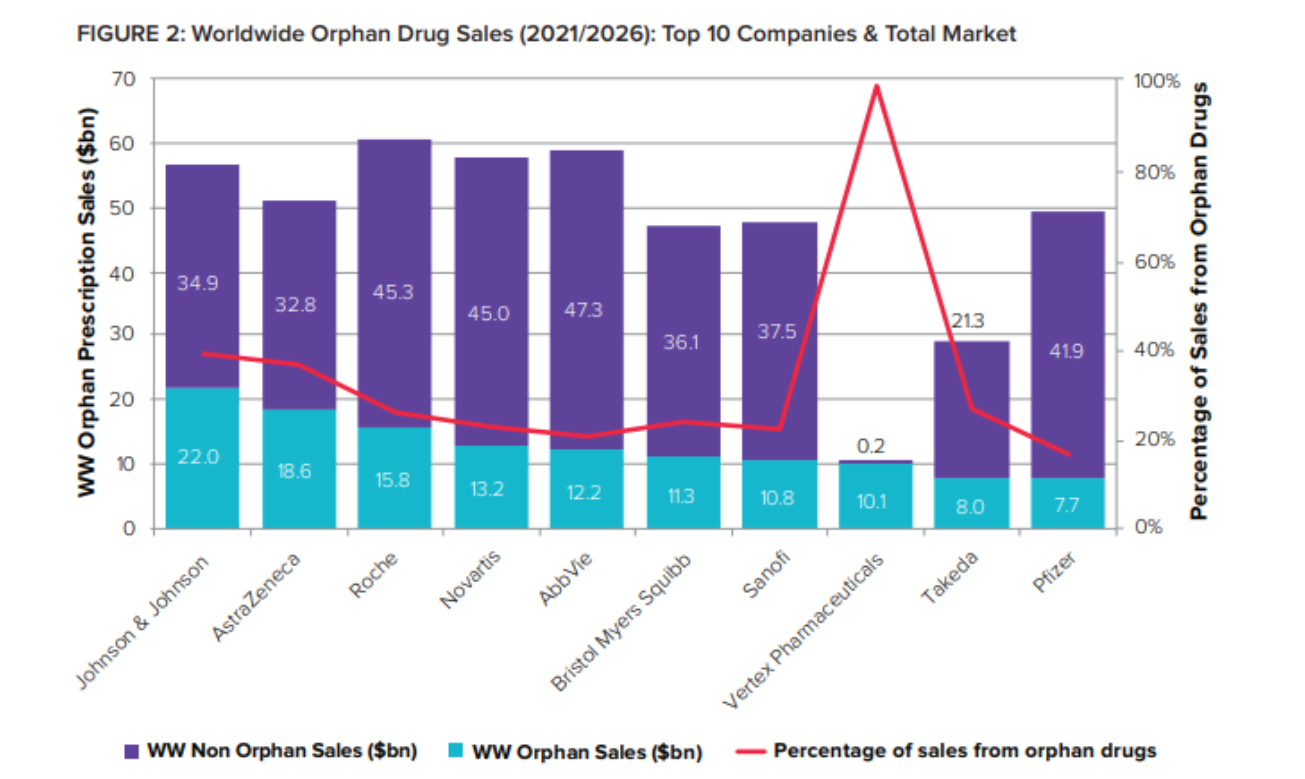
There has been a rapid acceleration of growth within the orphan drug market as science and the understanding of diseases have progressed, leading to orphan drugs no longer being niche.
As orphan drugs graduate from niche to mainstream and reach blockbuster heights, big pharma have adopted and embraced them – just as they did with biologics two decades ago.
The expansion of technology and the advancement of new tools have aided drug discovery and essentially brought orphan drugs into a new sphere within the drug market, renewing conversations for needed changes to legislation.
Revising legislation
There have been continued calls to reform orphan drug legislation in the US and the EU, particularly the 40-year-old Orphan Drug Act and the European Union’s 1999 Orphan Drug Regulation.
In the US, there are proposals to reform the FDA’s Accelerated Approval pathway, critical for orphan drugs that, as the report states, “include tighter oversight on post-marketing studies required to determine whether full approval is warranted for ‘accelerated’ products.”
The EU is reviewing its framework for orphan drugs after a report published in mid-2020 said that in some rare diseases, the market looked similar to the traditional medicines market.
Verdin and Hadjivasiliou say the calls for change are valid, though there are two ways one can view these appeals for modifications to legislation.
“Some things highlighted in the Report are the growth in the actual budgetary impact of drugs. Some could say that when you think of a rare disease that conjures up images of super-rare genetic conditions, rather than things like oncology, which are areas that attract a lot of interest, and often designations,” Verdin states.
“That’s part of a driving force around some of those voices making noises about revisiting that legislation – is it still fit for purpose on that basis?”
As the report notes, the challenge for payers and regulators is that orphan incentives are applied to drugs much larger than the US Orphan Drug Act or EU Orphan Regulation originally envisaged.
Alternatively, Verdin and Hadjivasiliou say, the counterargument is that the legislation aims to promote innovation and investment and generate positive technological outcomes.
Regarding oncology, for example, much has changed about the understanding of cancer since the Orphan Drug Act in the US was signed into law 40 years ago.
“Science has progressed considerably since the legislation was implemented in the 80s. There’s been a move towards oncology as a more detailed set of almost sub-diseases with genetic markers within those,” Hadjivasiliou states.
“It’s pretty well-known that cancer is not one disease. It’s a whole host of distinct diseases with similar manifestations that are distinctly important in terms of clinical practise and responses to drugs with certain mechanisms of action,” Verdin states.
Therefore, many people arguing for a change to legislation may perhaps view the legislation as not keeping with the current understanding of drugs available. Still, Verdin and Hadjivasiliou state the original purpose of incentivising innovation through this legislation has been fulfilled.
“The outcomes that we’re seeing now are perhaps not exactly as intended, but they were the broad spirit of what was trying to be achieved,” Hadjivasiliou states.
Positive projections
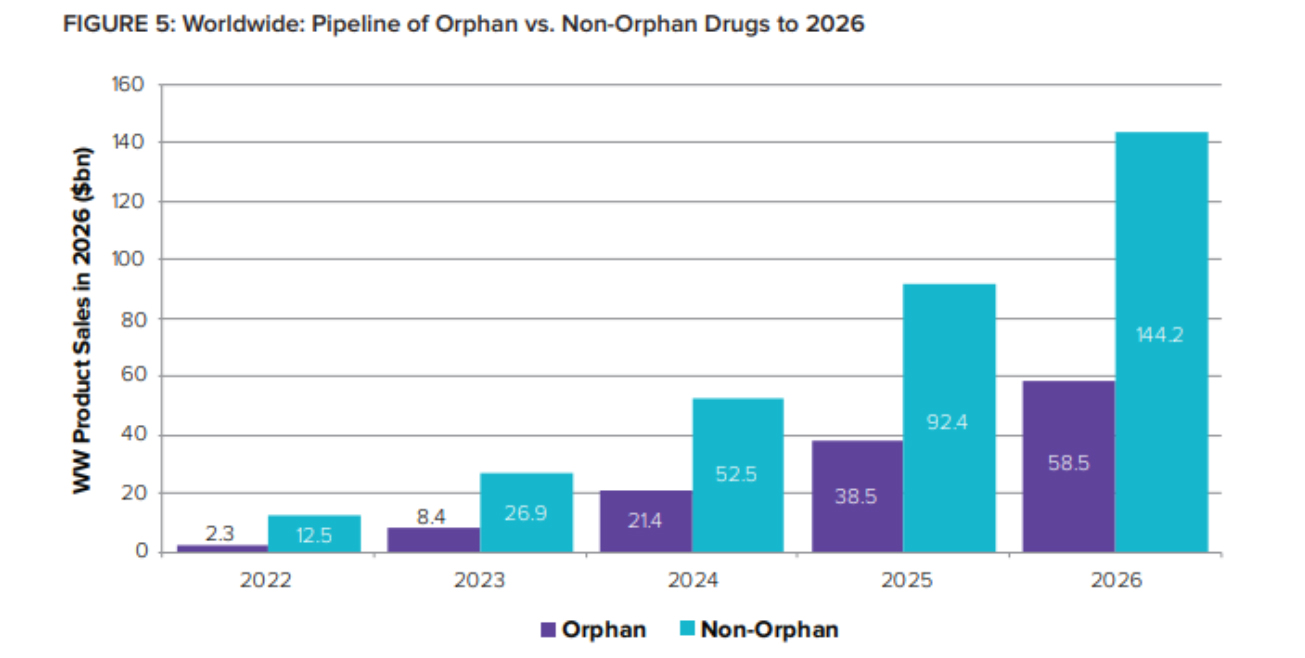
With innovation consistently improving and orphan drug sales projected to grow, the drug market has the potential to become more acceptable for the rare disease community.
“Because of the nature of the pharma industry and the time it takes to develop a drug successfully, or in many cases, unsuccessfully, that rate of change is a little bit slower. I think back over the various iterations of the Orphan Drug Report, and we’ve moved from a situation where pharma was very much interested in orphan drugs but perhaps hadn’t fully demonstrated their commercial return,” Hadjivasiliou states.
“It gradually became apparent that there is a significant commercial return here, and there’s an opportunity for mergers and acquisitions in this space. If you have a pipeline, you hope to see a return, and there are patients at the end of this who do see a benefit.”
Orphans’ share of global R&D pipeline value is expected to jump from 16% in 2022 to 29% just two years later – representing 2026 sales of over $58 billion.
Developing drugs is complex and expensive, often driving costs to be passed on to patients. However, with more pharma companies entering the orphan drug market, patients can be hopeful for additional therapeutics allowing for lower-cost options.
“Pharma has clearly demonstrated the ability to generate meaningful impact with new products that change people’s lives and have a material effect on one’s quality of life,” Verdin states.
“The key headline is that rare diseases are not a niche. It’s mainstream pharma and a big part of the pharma market. Developing drugs for rare diseases is also a breeding ground for next-generation therapeutics,” Verdin states.
“New sciences are coming through in this area. That’s quite an exciting space, and I think that’s something we’re going to see a lot of over the next few years,” Hadjivasiliou states.
About the interviewees
Dr. Paul Verdin is VP consulting and analytics at Evaluate. Verdin has a PhD in Neuropharmacology and leads Evaluate’s custom solutions business incorporating all aspects of forecasting and valuation analysis, opportunity identification and quantification, and industry trend and outlook assessment. With over 15 years of experience across research and consulting, he has worked with a wide range of client organisations in across therapy areas and across the pharma ecosystem, including startup biotech companies, suppliers and large pharma corporate strategy groups seeking insights and external perspectives to aid decision making.
Andreas Hadjivasiliou is the managing analyst at Evaluate. He is an experienced pharmaceutical industry analyst, currently engaged in driving process change and excellence within Evaluate’s Data Operations team. Most recently, Hadjivasiliou acted as a product owner leading remote and offshore teams delivering operational change. In previous roles, he helped design and deliver new data products for Evaluate, including insights on Orphan Drugs.
Acadia’s attempt to expand Nuplazid’s label into Alzheimer’s disease psychosis looks to have failed, following an FDA panel vote last Friday that went 9 to 3 against the idea. This was always a long shot afterthe complete response letter for the group’s previous applicationin the wider dementia-related psychosis setting; the Alzheimer’s submission relied heavily on a subgroup analysis and the FDA had already said it wanted another trial conducted. The company has yet to say whether this will happen; Alzheimer’s disease psychosis might have big commercial potential but investors are unlikely to want to see more money spent here. Either way, another CRL looks to be on the way, making negative symptoms of schizophrenia Acadia’s next label expansion bid; Nuplazid managed only a very narrow win in the first pivotal trialconducted here. Regarding the psychosis panel, the most supportive comments that Stifel analysts could muster were that the absence of negative surprises validated management’s credibility, though the logic that this is "incrementally positive" for the group’s next project, trofinetide in Rett syndrome, is pretty hard to follow. A filing for trofinetide is due mid-year, making acceptance from the FDA the next big event for Acadia.
Thousands of people in China's Macau lined up for Covid testing on Monday (June 20).
The world's biggest gambling hub reported dozens of locally transmitted cases over the weekend.
Most businesses have shut, but casinos will remain open.
Most of the territory's 600,000 residents have been told to stay at home and border restrictions have been tightened.
Shares of Macau casinos tumbled on Monday morning with some falling as much as 8%.
Macau's government relies on casinos for over 80% of its income.
Most of the population is employed directly or indirectly by the industry.
In a statement, Macau's chief executive Ho Iat Seng said the outbreak had come suddenly and was spreading rapidly.
Its source is still unknown.
Macau only has one public hospital and its services are already stretched on a daily basis.
The former Portuguese colony plans to test its entire population by Tuesday (June 21) as it adheres to China's strict zero-covid policy of attempting to eradicate all outbreaks.
Pfizer has agreed to invest 90.5 million Euros, or $95.24 million, to buy an 8.1% stake inFrench vaccine company Valnevaas the two companies revealed advancements in their partnership to stop Lyme disease.
The stake in Valneva, which is also working on a coronavirus vaccine, will be purchased for 9.49 Euros, or $9.99, per share, through a reserved capital increase.
The per-share purchase price was reached based on the average closing price of the "Company’s Shares on Euronext Paris during the 10 trading days preceding the date of the Equity Subscription Agreement," the companies said in press release. Shares in Valneva increased by 14.5% in early session trading to 9.09 euros.
Valneva is expected to use the proceeds from Pfizer’s $95 million investment to support its Phase 3 development contribution to the Lyme disease program.
It was announced on April 26, 2022, that Pfizer plans to initiate the Phase 3 study of Lyme disease vaccine candidate VLA15 in the third quarter of 2022.
Pfizer and Valneva also updated the terms of their collaboration and license agreement that they announced in April 2020 for VLA15.
Valneva will now fund 40% of the remaining shared development costs, an increase over the 30% from the initial agreement.
Pfizer will pay Valneva tiered royalties ranging from 14% to 22% compared to 19% in the prior agreement. The royalties will also be complemented by up to $100 million in milestones payable to Valneva based on cumulative sales.
"Pfizer's investment in Valneva highlights the quality of the work that we’ve done together over the past two years and is a strong recognition of Valneva's vaccine expertise," Valneva chief executive Thomas Lingelbach said in the press release. "This subscription agreement will contribute to our investment in the Phase 3 study while limiting the impact on our cash position."
The U.S.Food and Drug Administration has a handful of PDUFA dates over the next two weeks. Here's a closer look.
BMS's Breyanzi for R/R Large B-Cell Lymphoma
Bristol Myers Squibb has a target action date of June 24 for the supplemental Biologics License Application (sBLA) for Breyanzi for the treatment of adults with relapsed or refractory large B-cell lymphoma after failure of first-line therapy. The sBLA is built on data from the Phase III TRANSFORM trial.
Breyanzi (lisocabtagene maraleucel) is a CD19-directed chimeric antigen receptor (CAR) T-cell therapy. It is indicated for adults with r/r DLBCL after two or more lines of systemic therapy, including diffuse DLBCL not otherwise specified, high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma graded 3B.
The TRANSFORM study comparedthe drug as second-line therapy to the standard of care of salvage chemotherapy followed by high-dose chemotherapy plus autologous hematopoietic stem cell transplant. The data provided highly statistically significant and clinically meaningful improvements in event-free survival, complete responses and progression-free survival, and a positive trend in overall survival in patients with LBCL whose disease was primary refractory or relapsed within 12 months after first-line therapy.
TG Therapeutics' U2 for CLL and SLL
TG Therapeutics originally had a target action date of April 22 for a meeting of the FDA's Oncologic Drugs Advisory Committee (ODAC) to review its BLA and supplemental New Drug Application (sNDA) for the combination of ublituximab and Ukoniq (umbralisib), referred to as U2, for treatment of adults with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). The company announced that the FDA had extended the PDFUA date to June 25.
The submissions were based on data from the UNITY-CLL Phase III study of U2 compared to an active control arm of Obinutuzumab plus chlorambucil in patients with both treatment-naïve and relapsed or refractory CLL. The primary endpoint was superior progression-free survival (PFS) for U2 compared to the control arm, which it met, significantly prolonging the independent review committee assessed PFS versus control, 31.9 months versus 17.9 months, at a median follow-up of 36.7 months.
Then, on April 15, the company voluntarily withdrew the BLA/sNDAs based on recently updated overall survival (OS) data from the UNITY-CLL trial showing an increasing imbalance in OS. TG also voluntarily withdrew Ukoniq from sale for approved indications of adults with marginal zone lymphoma (MZL) who had received at least one previous anti-CD20-based regimen and adults with follicular lymphoma (FL) who had received at least three previous systemic therapies.
Spero's Tebipenem for Urinary Tract Infections
Spero Therapeutics has a target action dateof June 27 for its NDA for tebipenem HBr oral tablets for adults with complicated urinary tract infections (cUTI), including acute pyelonephritis caused by susceptible microorganisms. The drug has been granted Qualified Infectious Disease Product (QIDP), Fast Track, and Priority Review designations for these indications. The submission includes data from the Phase III ADAPT-PO trial, which hit the primary endpoint of demonstrating the drug was statistically non-inferior to intravenous ertapenem in treating these patients. The drug is being developed as the first oral carbapenem antibiotic for use in cUTI.
Merck's Pneumococcal Vaccine
Merck has a target action date of July 1 for its supplemental BLA for Vaxneuvance (Pneumococcal 15-valent Conjugate Vaccine) in infants and children, specifically for the prevention of invasive pneumococcal disease in children six weeks through 17 years of age. It is under Priority Review.
The vaccine consists of purified capsular polysaccharides from S. pneumoniae serotypes 1, 3, 4, 5, 6A, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F and 33F individually conjugated to CRM197 carrier protein. It is currently approved for adults 18 years and older for the prevention of invasive diseases caused by the S. pneumoniae serotypes contained in the vaccine.

 Dr. Paul Verdin is VP consulting and analytics at Evaluate. Verdin has a PhD in Neuropharmacology and leads Evaluate’s custom solutions business incorporating all aspects of forecasting and valuation analysis, opportunity identification and quantification, and industry trend and outlook assessment. With over 15 years of experience across research and consulting, he has worked with a wide range of client organisations in across therapy areas and across the pharma ecosystem, including startup biotech companies, suppliers and large pharma corporate strategy groups seeking insights and external perspectives to aid decision making.
Dr. Paul Verdin is VP consulting and analytics at Evaluate. Verdin has a PhD in Neuropharmacology and leads Evaluate’s custom solutions business incorporating all aspects of forecasting and valuation analysis, opportunity identification and quantification, and industry trend and outlook assessment. With over 15 years of experience across research and consulting, he has worked with a wide range of client organisations in across therapy areas and across the pharma ecosystem, including startup biotech companies, suppliers and large pharma corporate strategy groups seeking insights and external perspectives to aid decision making. Andreas Hadjivasiliou is the managing analyst at Evaluate. He is an experienced pharmaceutical industry analyst, currently engaged in driving process change and excellence within Evaluate’s Data Operations team. Most recently, Hadjivasiliou acted as a product owner leading remote and offshore teams delivering operational change. In previous roles, he helped design and deliver new data products for Evaluate, including insights on Orphan Drugs.
Andreas Hadjivasiliou is the managing analyst at Evaluate. He is an experienced pharmaceutical industry analyst, currently engaged in driving process change and excellence within Evaluate’s Data Operations team. Most recently, Hadjivasiliou acted as a product owner leading remote and offshore teams delivering operational change. In previous roles, he helped design and deliver new data products for Evaluate, including insights on Orphan Drugs.