Back in March, I wrote a post asking whether the GLP-1 “miracle” weight loss drugs—Wegovy, Ozempic, and the like—work primarily by suppressing appetite in the brain. That’s the conventional view. They quiet food noise and inhibit cravings, all by targeting the appropriate neurons in the brain. But is it right?

I care about this, of course, because I’m obsessed with the question of why we get fat to begin with. If these drugs can make us lose weight merely by getting us to eat less, that suggests that eating too much is why we get fat. I don’t buy it. Once upon a time I did, but I got over it.
For those unfamiliar with my work on obesity, here’s the context necessary to understand this issue, and how it relates to the GLP-1 drugs. (For those who are familiar, feel free to skip on down to the next section.)
A brief primer on the why we get fat question
Conventional thinking since the Second World War (but not before) has been that obesity is caused by taking in more calories than we expend. It’s an energy balance disorder, by this way of thinking. Simply put, we eat too much.
On one level that can seem obvious, but it fails to explain the singular observation that researchers thinking about obesity prior to the war had set out to explain. This is why they saw the etiology of obesity as a fascinating scientific challenge rather than just an abject lesson in the penalties of gluttony and sloth: Many people become obese eating no more food than people who stay effortlessly lean. Many people become obese, maybe most, while doing everything they can to resist it, including consciously restricting how much they eat. So what explains their obesity?1
Answering that question resulted in an alternative explanation/hypothesis: we get fat not because of how much we eat, but because of what our bodies do with what we eat. Some bodies prioritize the storage of fat over the use of fat for fuel and so accumulate fat to excess. Others, the lean ones, don’t.
This is a fuel-partitioning hypothesis of obesity: the dysregulation is not with appetite and how much we eat, but with this partitioning of fat between fuel use (oxidation, in the lingo) and storage. And because that partitioning is determined primarily by hormonal responses to the food we eat, and by the central nervous system, this hypothesis is a neuroendocrine hypothesis. (And because the dominant hormone promoting fat storage is insulin, the dietary implication of this thinking is the carbohydrate-insulin model, or CIM.)
If this hypothesis is right, then obesity researchers have been misinterpreting their evidence and data for going on 80 years. That’s a hard one to accept, but the evidence itself supports it.
For those who would like to learn more about this fuel-partitioning hypothesis, see the essay I wrote for STAT News in 2021, which describes its history and implications. The science is discussed technically in this extensive review article in Obesity Reviews published last summer.
By this perspective, those of us who struggle with our weight do so because of this physiological prioritization of fat storage over fat oxidation. Our bodies act as though they’re food deprived, despite the excessive amount of fuel (fat) that’s stored in our fat tissue. If we’re hungrier then we would be otherwise—if we’re always fighting off cravings and food noise, as this is now described—this is the reason why.
How do the GLP-1 drugs inform this alternative model of obesity?
If this fuel-partitioning model of obesity is correct, then it’s quite likely that the GLP-1 drugs work by reversing this fat-trapping process, unlocking fat stores. mobilizing the fat that’s been stored, and shifting the body into a fat-burning state. This shift reduces appetite indirectly—because the body is now fueling itself from within, from its own fat stores. It’s finally making use of all that fat that’s been acting as a burden rather than a storage depot.
Take the drug, by this scenario, shift the partitioning of fat from storage to oxidation, and now the internal hunger vanishes as does all the associated food noise and cravings.2 Appetite drops and food noise quiets not because the brain is tricked into feeling full, as the conventional wisdom has it, but because the body’s energy needs are being met. The brain doesn’t respond to the weight loss as though the body is being starved of food, because it’s not. (The same argument holds for the inhibition of appetite that accompanies ketogenic diets.)
As I’ve noted in earlier posts, researchers rarely do the experiments necessary to distinguish these competing mechanisms because they assume the answer is already known based on their assumptions about obesity itself. If we get fat because we eat too much and people on the drugs both eat less and lose weight, it’s a natural assumption: the drugs worked to make people eat less, and the eating less causes the weight loss. Simple. But is it right?
Occasionally, however, researchers do studies that provide evidence that allows us to test these competing hypotheses. That’s what I wrote about in March, both the patient experience on the GLP-1 drugs and a study that had recently been published. Now we have another study published this week in Cell Metabolism that adds yet more evidence. The researchers—mostly from the Pennington Biomedical Research Center in Louisiana and from Eli Lilly, which makes the drug and funded the study—are investigating the actions of Tirzepatide, which is the active ingredient in Mounjaro.
Their paper is a report of the drug’s effects in both obese mice and obese humans. What makes it useful for our purposes is that the researchers went beyond the usual appetitive phenomena they measure in these studies—food intake, hunger and satiety, food noise and cravings. They also measured physiological parameters that could shed light on what these drugs are doing to this fuel partitioning process: Not just their effect on how much we eat, but their effect on what we do with what we eat, the competing concepts in this why-we-get-fat question.
The design: pair-feeding in mice, free-feeding in humans
The mouse experiment was relatively simple. The mice were particularly susceptible to getting fat on the diet they fed them (diet-induced obese (DIO) mice, in the lingo), but the researchers only allowed them only half their preferred daily ration of calories. So these animals were, quite literally, semi-starved.
The mice were then divided into three groups:
Mice that were given injections of Tirzepatide
A placebo-treated group that was pair-fed to the Tirzepatide group. This means that how much these mice were allowed to eat on any given day was determined by how much the Tirzepatide-treated mice did eat.
A placebo-treated group without the pair-feeding. So these animals got the 50% calorie-restricted diet but they could eat all of it (which I’m willing to bet they did).
The key comparison is between the Tirzepatide-treated mice and the pair-fed mice that got only a placebo. These animals consumed the identical number of calories—at most, half of what they’d prefer to eat—so that controlled for the effect of the drug on appetite. If the drug's only function was appetite suppression, then both groups should have lost the same amount of weight. But that’s not what happened, as I’ll discuss.
In the human trial, the design was simpler still: 55 participants with obesity were randomized to receive either Tirzepatide or a placebo for 18 weeks. Unlike the mice, the human participants were allowed to eat as much as they wanted (albeit with what appears to be the usual guidance to restrict calories and at least try to lose weight that way3).
In both studies, the researchers used a technique called indirect calorimetry to measure two key outcomes: How much energy the participants (mice or humans) expended. And a variable called the Respiratory Exchange Ratio (RER).
RER requires a little explanation. Technically, it’s the ratio of carbon dioxide exhaled to oxygen consumed. But this ratio then tells us what fuel the body is burning. An RER close to 1.0 means mostly carbohydrates; closer to 0.7 indicates mostly fat.4 Shifts in RER reveal shifts in metabolic strategy: is the body burning carbs (glucose) for fuel or burning fat and, in this case, how does that change with the drug?
RER in the context of the pair-feeding and the semi-starvation tells us how the drug affects this fuel-partitioning question independent of how much food is eaten.
The results: something more (or other) than appetite suppression
In the mouse study, Tirzepatide-treated mice lost significantly more weight than the pair-fed mice, despite both groups consuming identical calories. That finding is worth the price of admission right there.
But the Tirzepatide-treated mice also had a lower RER, indicating greater fat oxidation, and they expended more energy—suggesting that the drug blunted the normal drop in energy expenditure that comes with calorie restriction, a phenomenon known as metabolic adaptation.
That’s important. When animals or people cut calories, they typically expend fewer calories. We can think of it as a biological defense mechanism against starvation or as the body burning only the limited fuel available. Tirzepatide attenuated that reduction in energy expenditure in these mice, allowing them to continue burning more energy than their calorie-matched peers.
Here’s what that looked like in all the parameters the researchers measured. The tirzepatide-treated mice are the blue lines; the pair-fed mice are the green-lines and the placebo group without pair-feeding is the grey lines. (Notice that the green line and the blue line overlap in the first chart, A, showing calorie intake. This perfect overlap is the pair-feeding.)
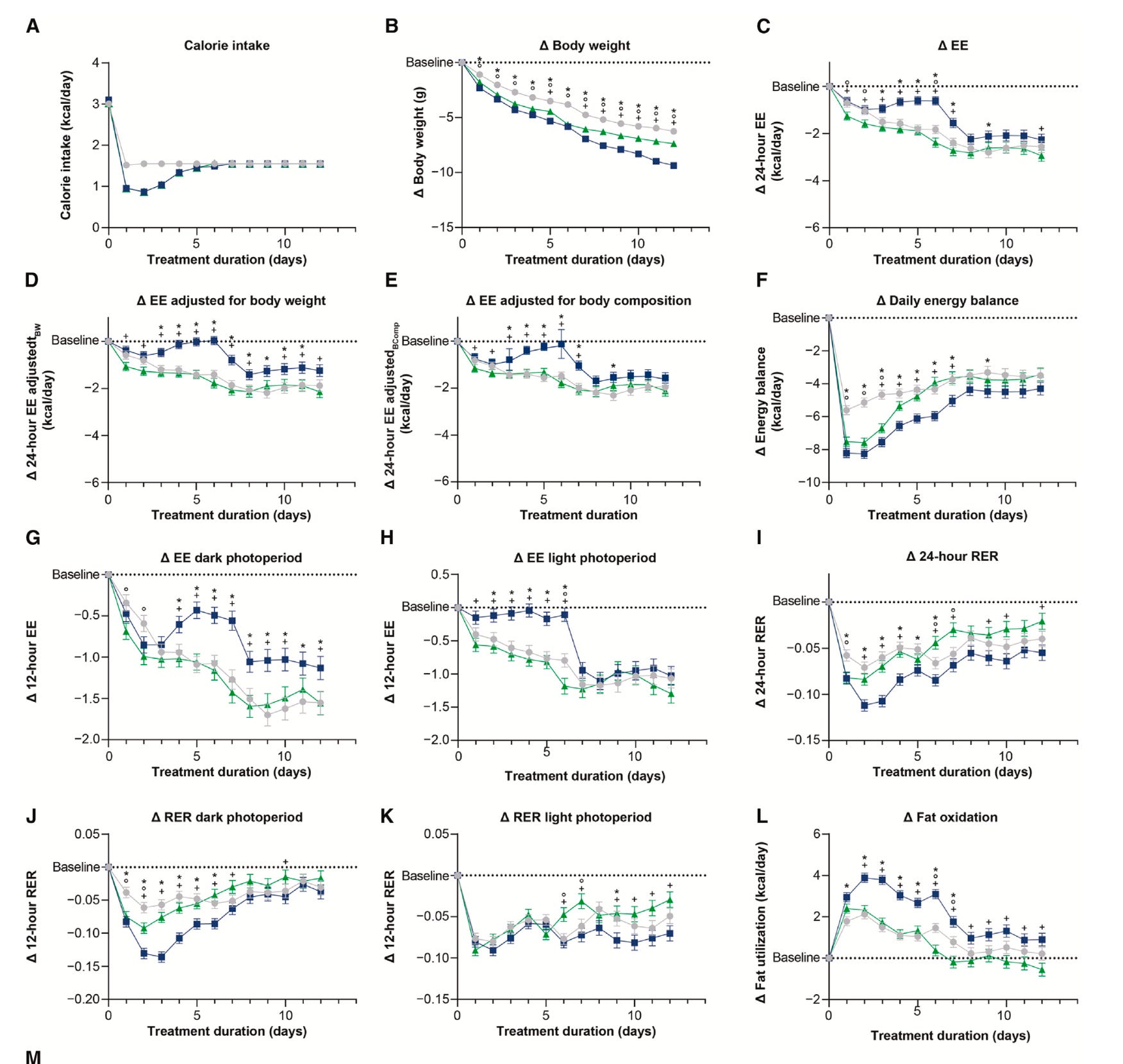
In humans, the results were less dramatic but pointed in the same direction. Participants on Tirzepatide burned more fat—RER was lower—than those on placebo. But the investigators observed no significant difference in energy expenditure, or at least not after they adjusted for the changes in body composition that came with taking the drug.
An important point, though, is that they only measured RER after body weights had plateaued, not during the active weight-loss phase, when metabolic adaptation is most evident. The authors themselves note this limitation, and it may explain why they didn’t see the same anti-adaptive effect in people as in mice. The adaptation might have occurred earlier, then receded by the time measurements were taken.
The investigators didn’t offer a mechanism for any of these observations—i.e., why a drug that reduces appetite would also promote fat burning and (if you’re a mouse, at least) allow you to expend more energy than you might otherwise. The fuel partitioning perspective very much does.
The fuel-partitioning lens
In short, all of this is consistent with the predictions of the fuel-partitioning hypothesis of obesity.
Remember, from the fuel-partitioning perspective, Tirzepatide’s primary effect is not to suppress appetite directly, but to liberate stored fat and, in the process, induce the rest of the body to prioritize that fat as a fuel source. To use the language of diet book doctors, the drug makes the body shift into a fat-burning mode (as shown by the drop in RER), which then:
Feeds the system from within, making the stored fat available for oxidation, which
Suppresses appetite indirectly, and
Attenuates the usual energy-conserving adaptations that go with calorie-restricted diets and weight loss.
The simplest way to think about it: People (and mice) on these drugs don’t act metabolically like they’re starving, because they’re not. Their tissues are being fueled by their own fat reserves. It’s the drug that allows this to happen. As those reserves diminish and weight plateaus, hunger returns, and energy expenditure once again aligns with actual food intake rather than with the availability of that excess energy from the fat tissue.
Do the researchers discuss this possibility when interpreting their observations?
Short answer: no. Whether they’re unaware of the fuel-partitioning model or simply unconvinced by it—mostly the first, I’m assuming, but that could be wishful thinking—they don’t mention it.
Why mostly the first? Because one of the lead authors on the study was Eric Ravussin of Pennington, and Ravussin has been a co-author on several papers directly engaging with this hypothesis. (This one, for instance, just last summer.) Ravussin is a highly respected obesity researcher. I’ve known him since I interviewed him for my first book in the early 2000s. I worked with him in my days with the Nutrition Science Initiative (NuSi), when he was a principal investigator on a NuSI-funded pilot study designed to address the experimental issues with testing these hypotheses.
I had recruited Ravussin to that experiment, because I found him to be one of the more thoughtful and open-minded researchers in the field. I suspect, though, that in these drug studies, his focus is confined to the drug’s immediate effects, not their broader implication for studying obesity. Worst case scenario: he doesn’t believe, as I do, that good science requires interpreting evidence in the light of all viable hypotheses, not just the hypothesis you prefer.
If obesity is better understood as a problem of fat trapping, a neuroendocrine fuel-partitioning disorder rather than an energy balance problem, that has profound implications for not just the treatment of obesity but for its prevention. As for the GLP-1 drugs, they may indeed work primarily by restoring access to that trapped fat, not by shutting down hunger signals in the brain.
Gary Taubes is an investigative science and health journalist, author of Rethinking Diabetes (2024), The Case for Keto (2020), The Case Against Sugar (2016), Why We Get Fat (2011), and Good Calories, Bad Calories (2007), published as The Diet Delusion in the UK.https://uncertaintyprinciples.substack.com/p/why-do-we-lose-weight-on-glp-1-drugs