Agios Pharmaceuticals, Inc.. (NASDAQ: AGIO), a leader in the field of cellular metabolism to treat genetically defined diseases, today announced the closing of the sale of its commercial, clinical and research-stage oncology portfolio to Servier Pharmaceuticals, LLC, an independent global pharmaceutical company. The transaction was approved by Agios shareholders on March 25, 2021.
In consideration for its oncology portfolio, Agios received from Servier $1.8 billion in upfront cash and is eligible to receive an additional $200 million in a potential future milestone payment for vorasidenib, as well as 5% royalties on U.S. net sales of TIBSOVO® (ivosidenib tablets) from sales after the closing through loss of exclusivity and 15% royalties on U.S. net sales of vorasidenib from the first commercial sale through loss of exclusivity.
In addition, Agios today announced that it has entered into a definitive agreement with Bristol-Myers Squibb Company (BMS) to repurchase 7,121,658 shares of Agios common stock held by BMS and its affiliates for an aggregate purchase price of $344.5 million, or $48.3785 per share, using the proceeds from the sale of the oncology business. As previously disclosed, the Agios board of directors authorized the company to repurchase up to $1.2 billion of its outstanding shares, using the proceeds from the sale of the oncology business. Following completion of the repurchase of shares from BMS, Agios expects to conduct the remaining $855.5 million of share repurchases over the next 12-18 months, including executing a meaningful portion of the planned repurchases by year-end through a combination of 10b5-1 plans and open market purchases.
With a singular focus on growing the company’s genetically defined disease clinical and research pipeline, Agios anticipates significant key milestones in 2021, including filing for regulatory approval for mitapivat in adults with PK deficiency in both the U.S. and EU; initiating two Phase 3 studies of mitapivat in transfusion dependent and non-transfusion dependent thalassemia; initiating a Phase 2/3 study of mitapivat in sickle cell disease; presenting the first data from the healthy volunteer study of AG-946, the next generation PKR activator; and prioritizing new indications for PKR and pyruvate kinase M2 (PKM2) activator clinical development. In addition, Agios will explore all options to maximize the patient impact and value of mitapivat globally, including strategic transactions.
Aytu BioPharma, Inc. (NASDAQ:AYTU) a specialty pharmaceutical company focused on commercializing novel therapeutics and consumer healthcare products, today announced the signing of an agreement with Acerus Pharmaceuticals Corporation (ASP)(ASPCF) whereby Acerus will acquire all remaining rights to Natesto in the United States from Aytu. In consideration, Aytu will receive $7.5M in cash from Acerus, which is payable in $250,000 monthly payments over 30 months. Additionally, Acerus will assume all product responsibilities associated with Natesto following the April 1, 2021 effective date. Aytu will provide transition support to Acerus over a 120-day transition period.
First and only approved therapy in the United States for patients with PH-ILD, a serious, life-threatening disease with potentially more than 30,000 patients in need
FDA approval based on data from the INCREASE clinical trial
PH-ILD is the second FDA-approved indication for Tyvaso, which was initially approved for the treatment of pulmonary arterial hypertension
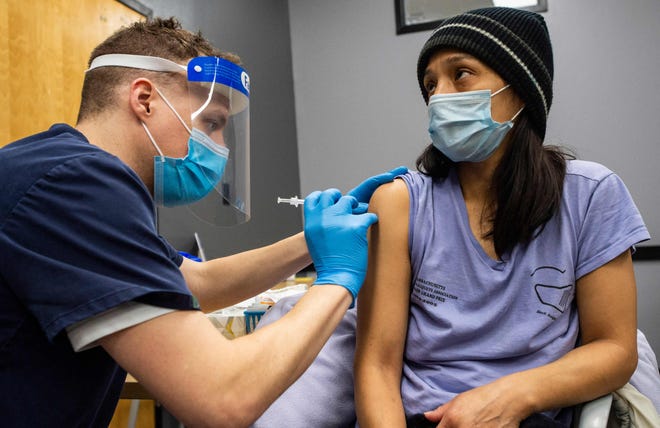
Six months after getting a second dose of the Pfizer-BioNTech vaccine as part of a 46,000-person clinical trial, volunteers remained more than 90% protected against symptomatic COVID-19 and even better protected against severe disease, a new company study found.
Out of 927 trial participants who fell ill with COVID-19 more than a week after their second dose, only 77 had received the active vaccine, compared with 850 who got a placebo.
There were no serious safety concerns seen among the 12,000 volunteers who are least least six months past their second dose, according to the newly released findings. Many, however, did have typical, short-term side effects like fatigue and sore arms.
The new data is likely sufficient for the vaccine to meet the criteria set by the U.S. Food and Drug Administration for full approval.
All three COVID-19 vaccines authorized so far – from Pfizer-BioNTech, Moderna and Johnson & Johnson – are being distributed under Emergency Use Authorizations rather than full FDA Biologics Licenses, because they did not have long-term safety and effectiveness data.
The FDA allowed the companies to present only two months of data so they could get the COVID-19 shots to the public faster during a global emergency. The agency has said it would consider issuing a full license once a vaccine has a track record of at least six months – the same requirement as for vaccines that prevent other infectious diseases.
Pfizer and its German collaborator BioNTech, whose vaccine has been administered to nearly 77 million Americans, are expected to submit their application for full licensure to the FDA some time this month.
"These data confirm the favorable efficacy and safety profile of our vaccine and position us to submit a Biologics License Application to the U.S. FDA,” Pfizer CEO Albert Bourla said in a prepared statement.
Others said the data suggests the vaccine deserves full approval. "It seems pretty safe" to assume that the agency will fully license the vaccine, said Luciana Borio, a biotech executive and former FDA chief scientist.
"It's really wonderful news that these vaccines continue to progress very rapidly toward licensure," she said.
Full licensure, she said, would be "one less reason for the skeptics to decline vaccination."
But the general public will notice very little difference, said Jacob Sherkow, a law professor at the University of Illinois College of Law.
The main advantage of licensure, he said, is to provide security to the company that its authorization won't expire. A Biologics License usually provides a company 12 years of exclusivity for its product.
Emergency Use Authorizations, by contrast, are temporary, Sherkow said. "If the FDA grants an EUA, it can yank it any time it wants for any reason or no reason." It would also expire when the "emergency" is over.
"I think (licensure) will give the company some solace that they're not going to be victims of their own success," he said. "If you have a very effective vaccine and the vaccine ends the pandemic, then you've ended the emergency that authorized the EUA and you're not authorized anymore. The BLA guarantees that you get to stay on the market indefinitely."
The FDA will likely move quickly to consider full licensure, Borio said, but noted that staffers are busy with other requests, too.
Pfizer and BioNTech said Wednesday they would ask the FDA for permission to provide their vaccine to adolescents ages 12-15. The vaccine is already authorized for use in older adolescents, and new data suggests it's just as safe and totally protective in younger ones.
The agency is also expected to be asked within the next few days to review an application for authorization from AstraZeneca and Oxford University for the vaccine they've co-developed.
In their latest study Pfizer and BioNTech also showed that their vaccine is effective against a virus variant called B.1.351, first identified in South Africa.
About 800 of the trial participants were from South Africa and nine of them developed COVID-19, all in the placebo group. The companies sequenced the genes of all nine of those infected and found that six belonged to the B.1.351 variant.
“These data also provide the first clinical results that a vaccine can effectively protect against currently circulating variants, a critical factor to reach herd immunity and end this pandemic for the global population,” Ugur Sahin, CEO and co-founder of BioNTech, said in a prepared statement.
The vaccine also appeared to be extremely effective against nearly all cases of severe disease across all trial participants.
The FDA and Centers for Disease Control and Prevention have slightly different definitions for "severe disease." Under the FDA's definition, there were 21 cases of severe disease, 20 of whom received a placebo. Under CDC's definition, 32 trial participants developed severe disease, all of them in the placebo group, according to the study.
Pfizer and BioNTech plan to submit the new data to a peer-reviewed journal for review and publication.
They will continue to follow trial participants for two years to ensure continued safety and effectiveness and to begin to answer one of the last major outstanding questions about COVID-19 vaccines: how long they will remain effective.
Brazilian hospital chain Care has filed for an initial public offering of roughly 790.5 million reais ($139.42 million), amid a dealmaking boom in Brazil's healthcare sector, according to a securities filing on Wednesday.
Care's plans for an IPO follows a raft of share offerings by hospital operators. Rede D'Or successfully concluded in December a 11.4 billion reais IPO and rivals such as Kora Saude Participacoes and Dasa are expected to conclude share offerings in the coming weeks.
Rising demand for services and an aging population have fueled deals in Brazil's healthcare sector, as Reuters reported earlier.
Hospital Care and its shareholders plan to sell at least 31 million shares between 22.50 reais and 28.50 reais. If overallotments are sold, the offering may increase by 35%. The price will be set on April 20.
The hospital operator has among its shareholders Brazilian private equity firm Crescera and Elie Horn, the founder of homebuilder Cyrela.
It operates 11 hospital, with 1,206 beds, and also sells healthcare plans, in a business model similar to Hapvida and Notre Dame Intermedica.
Itau BBA, BTG Pactual, Bank of America, XP, Safra and UBS BB will manage the offering.
Johnson & Johnson said one batch of its new Covid-19 vaccine didn't meet quality standards at a contract manufacturer, and the doses weren't distributed.
J&J said Wednesday the batch never advanced to the filling and finish stages of its manufacturing process, and that the quality lapse wouldn't affect its ability to supply the U.S. with 100 million doses by the end of May.
J&J didn't disclose the nature of the quality lapse or how many doses were affected, but said it arose from quality checks during the start-up phase of manufacturing. The company said it shared information about the issue with the U.S. Food and Drug Administration.
The FDA is investigating, according to a person familiar with the matter.
The New York Times reported Wednesday that the J&J doses were ruined due to an accidental mix-up of ingredients at Emergent BioSolutions Inc., the contract manufacturer working with J&J. The Times reported that about 15 million doses were ruined.
Emergent declined to comment.
J&J's vaccine was the third to be authorized for use against Covid-19, after shots from Pfizer Inc. and its partner BioNTech SE and from Moderna Inc. Health authorities especially welcomed the addition of the J&J vaccine because it requires just one dose and is easier to store.
Supplies in the U.S. were expected to increase as J&J's manufacturing network ramped up production, accelerating a mass vaccination campaign that has been gaining steam.
J&J said it is providing additional experts in manufacturing, technical operations and quality to be on-site at Emergent to oversee all manufacturing of the J&J vaccine there.
Emergent, a contract manufacturer based in Gaithersburg, Md., has been making the main ingredient for J&J's vaccine at an Emergent plant in Baltimore.
At the same plant, Emergent also makes the main ingredient for AstraZeneca PLC's Covid-19 vaccine, which hasn't yet been authorized for use in the U.S.
J&J's own plant in the Netherlands has been making the main ingredient for the initial U.S. supply of its vaccine -- including the nearly four million doses that were distributed immediately after it was authorized in late February.
J&J said it was able to meet a target of delivering a total of 20 million vaccine doses by the end of March. The company said it expects to deliver an additional 24 million doses in April, and plans to have delivered a total of 100 million by the end of May.
COVID-19 can cause acute respiratory distress syndrome (ARDS), leading to death in a significant number of individuals. Evidence of a strong role of the innate immune system is accumulating, but the precise cells and mechanism involved remain unclear. In this study, we investigated the links between circulating innate phagocyte phenotype and functions and severity in COVID-19 patients. Eighty-four consecutive patients were included, 44 of which were in intensive care units (ICU). We performed an in-depth phenotyping of neutrophil and monocyte subpopulations and measured soluble activation markers in plasma. Additionally, myeloid cell functions (phagocytosis, oxidative burst, and NETosis) were evaluated on fresh cells from patients. Resulting parameters were linked to disease severity and prognosis. Both ICU and non-ICU patients had circulating neutrophils and monocytes with an activated phenotype, as well as elevated concentrations of soluble activation markers (calprotectin, myeloperoxidase, neutrophil extracellular traps, MMP9, sCD14) in their plasma. ICU patients were characterized by increased CD10low CD13low immature neutrophils, LOX-1+ and CCR5+ immunosuppressive neutrophils, and HLA-DRlow CD14low downregulated monocytes. Markers of immature and immunosuppressive neutrophils were strongly associated with severity and poor outcome. Moreover, neutrophils and monocytes of ICU patients had impaired antimicrobial functions, which correlated with organ dysfunction, severe infections, and mortality. Our study reveals a marked dysregulation of innate immunity in COVID-19 patients, which was correlated with severity and prognosis. Together, our results strongly argue in favor of a pivotal role of innate immunity in COVID-19 severe infections and pleads for targeted therapeutic options.
Competing Interest Statement
The authors have declared no competing interest.
Funding Statement
This work was funded by the French National Research Agency (ANR) RA-COVID-19 V5 grant under the name COVINNATE